CNrun example
Supplementary material for the PLoS paper “Competition-based model of pheromone component ratio detection in the moth.” by Zavada A et al.
Note: the experimental setup described here uses CNrun 1.x, not the newer, Lua-powered version 2 (for that, check out this annotated script).
Experimental design
We used minimalistic, simplest conceivable neuronal network capable of detecting a certain target ratio of intensities of input components, by means of competition.
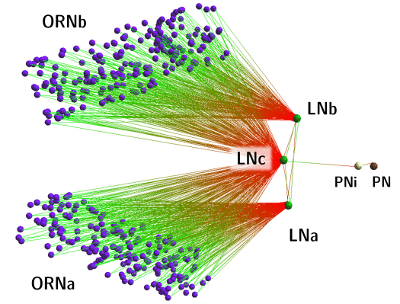
The model can be created:
- In a tool such as neuroConstruct which generates a NeuroML
network topology file (this file was
actually used in our experiments; note however, that each of
the clouds of ORNs are represented by a single Poisson
oscillator). Use
load_nml m.nml
to load the model. - Directly in cnrun with the following commands:
new_model ratiocoding add_neuron Poisson ORNa add_neuron Poisson ORNb add_neuron HH LNa add_neuron HH LNb add_neuron HH LNz add_neuron HH PNi add_neuron HH PN add_synapse AB ORNa LNa .1 add_synapse AB ORNa LNz .1 add_synapse AB ORNb LNz .1 add_synapse AB ORNb LNb .1 # transverse add_synapse AB LNa LNb .1 add_synapse AB LNb LNa .1 # inbound add_synapse AB LNa LNz .1 add_synapse AB LNb LNz .1 # outbound add_synapse AB LNz LNa .1 add_synapse AB LNz LNb .1
The model topology is now loaded in CNrun.
To actually run the simulation, you will need stimulation protocol files, and a CNrun script to properly set up all unit parameters. These files can be found in
$prefix/share/aghermann/ratiocodingafter you have donemake install(or installed the .deb package); for your convenience, they are also available in this file. In the directory where you have the contents of setup tarball extracted, execute thebatchshell script, like so:$ ./batch 5 1.5
After 5 trials times 6 times a couple minutes per trial, you will have in each of the six newly created trial directories some .varx files, which record the E of so named neurons; these can be plotted with
rational-plot-var. Similarly, the matrix data can be visulaised usingrational-plot-sdf-static(like those shown in fig. 4) or, in a fancier manner, usingrational-plot-sdf-interactive.
The
batchscript usesvarfold(part of cnrun package) to ‘fold’ the spike density function values into a matrix, which is then used to calculate a cost function for a given trial.